






The world's largest quantum chemistry dataset to empower new materials design and drug discovery
24/11/2022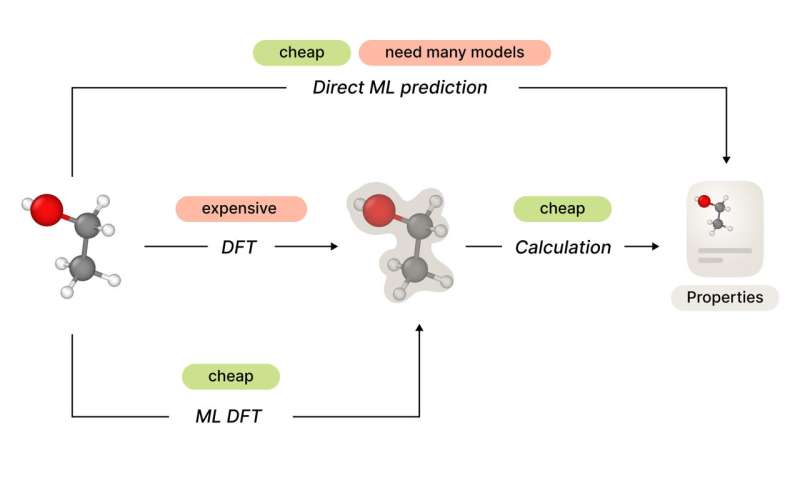
Figure 1. Different approaches to predict molecular properties. Credit: Artur Kadurin
Predicting the properties of anobject is a most natural task for machine learning (ML) algorithms, and molecules or crystals are not an exception. Every drug discovery or materials design pipeline depends on the ability to predict a future product's physical and chemical properties.
However, in contrast to more conventional domains of ML, such as images or texts, it is much more expensive in both money and time to validate the work of your models. To assess the quality, you must go to the wet lab to synthesize the structure and then perform real-world experiments to evaluate every single property. In addition to that, to train a machine learning model to predict molecular properties, you need access to relevant data for every property of interest, while the quality will depend on the size and diversity of your dataset.
A more general approach is to try to predict all the properties of the atomic system ab initio. Luckily, there is a fundamental theory behind the processes occurring on the quantum level. The Schrödinger equation allows us to explicitly calculate what is going on between atoms and electrons. For scientists, it means that we can simulate the behavior of a molecule or material and explicitly calculate its properties, at least in theory.
In practice, the amount of calculations needed for a precise solution of the Schrödinger equation grows exponentially with the number of electrons. However, there exist a wide variety of numerical methods that solve it on different levels of precision. These methods comprise a hierarchy that trades off accuracy against computational cost. Density functional theory (DFT) provides us with reasonably precise methods with feasible computation costs for systems of dozens of atoms.
Deep learning for quantum chemistry
Recent advances in deep learning (DL), especially in graph convolution networks, opened a whole new field of research—neural networks for quantum chemistry. Instead of predicting a specific property of a molecular structure, these methods are aiming to assess molecular conformation—the 3D arrangement of the atoms in a molecule, by predicting its quantum properties.
In particular, there are a number of papers focused on the substitution of computationally expensive DFT calculation with relevantly cheap neural network solutions. The vast majority of these works are limited to experiments performed only on a few or even single structures. It restricts the generalization and questions the applicability of these models to real-world problems.
nablaDFT dataset
On the path to solving the problem of access to suitable data, we at the DL in Life Sciences research group from AIRI, Artificial Intelligence Research Institute, decided to compute and share the biggest (so far) quantum chemistry dataset calculated on the DFT level of theory. The research was published in Physical Chemistry Chemical Physics and performed in collaboration with scientists from the Skolkovo Institute of Science and Technology and the St. Petersburg Department of Steklov Mathematical Institute. Together with the data we reimplement and evaluate several state-of-the-art neural network models on two common tasks: prediction of potential energy (a) and DFT Hamiltonian (b) for a given molecular conformation.
The dataset available via GitHub contains over 5 million conformations for over 1 million drug-like molecules together with quantum properties such as conformational energy, DFT Hamiltonian matrix, wave functions, and many others. It takes about 5 min of CPU time on average for a single conformation computation, which sums up to about 50 years of CPU time for the whole dataset.
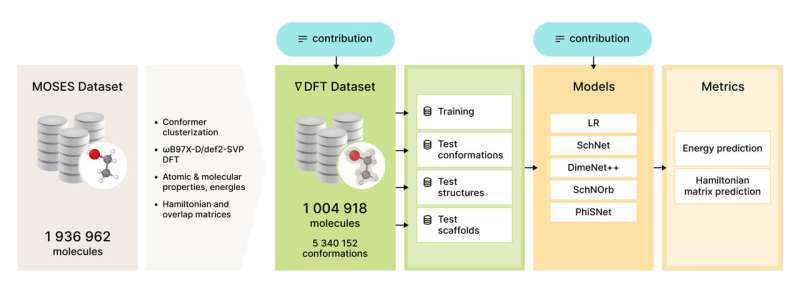
Figure 2. Overview of the nablaDFT dataset and benchmark contribution. Credit: Artur Kadurin
Benchmark results
To benchmark models in different settings we divide the test set into three subsets:
- Molecular conformations for structures presented in the training set.
Molecular conformations for structures not presented in the training set.
Molecular conformations for structures with scaffolds were not presented in the training set.
Conclusion
Though it remains a challenge to obtain models that are close to chemical accuracy, our experimental evidence shows that larger datasets lead to better ML models.
While we plan to keep replenishing the already collected dataset in order to contribute to the development of artificial intelligence technologies, we would like to invite the community to contribute to the benchmark by evaluating novel models on the proposed dataset.
This story is part of Science X Dialog, where researchers can report findings from their published research articles. Visit this page for information about ScienceX Dialog and how to participate.
Source: https://bit.ly/3UX4rMf, via Phys.Org.